Research
New Medicare Benefits Schedule (MBS) item for faecal calprotectin testing
From 1 November 2025 patients diagnosed with IBD who are symptomatic will be able to access a Medicare benefit for…

THE INNOVATE STUDY: TESTING A NEW DIET THERAPY FOR ULCERATIVE COLITIS
Monash University researchers are studying a new diet as a treatment for individuals with flaring ulcerative colitis.

Identifying the most important future research questions for perianal Crohn’s disease: a prioritisation survey
Researchers are seeking to identify the top priorities for people living with perianal Crohn’s Disease. Please answer this short survey…

IBD and diet – patient questionnaire
IBD and diet – patient questionnaire Monash Health Gastroenterology researchers are conducting a survey on the effects of diet on…

Collective 202 Study
Patients can find locations in Australia by visiting Study Details | VE202 in Patients with Mild-to-Moderate Ulcerative Colitis | ClinicalTrials.gov

Global Perianal Crohn’s Disease – research survey
Researchers are seeking to identify the top priorities for people living with perianal Crohn’s Disease. We are inviting people to participate in a brief survey to highlight the most crucial areas for future research. This will guide future studies, ensuring they address the most key issues raised by those with lived or professional experience of perianal Crohn’s disease.

Study: Understanding the perceptions, attitudes, and use of protein supplements amongst adults with ulcerative colitis
Calling all adults with UC, you are invited to participate in a research study conducted by a collaborative group of…

RACGP article: ‘GPs being left out of patients’ IBD treatment’
The Royal Australian College of General Practitioners (RACGP) recently covered CCA’s recommendations and findings from the IBD Paediatric Quality of…
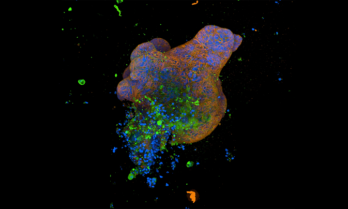
Cambridge University lab grown “mini guts” study
"Cambridge scientists have grown ‘mini-guts’ in the lab to help understand Crohn’s disease, showing that ‘switches’ that modify DNA in gut cells play an important role in the disease and how it presents in patients. These could in future be used to identify the best treatment for an individual patient, allowing for more precise and personalised treatments."

10 news & Garvan Institute of Medical Research segment
A segment on 10 News First that aired yesterday on the Garvan Institute of Medical Research autoimmune disease research project.

Robot surgery for Inflammatory Bowel Disease
Treatment for IBD progresses every year, with the use of biologics reducing the rate of surgery. However, in IBD that does not respond to…

Evaluation of a person-centred communication aid tool for people with Inflammatory Bowel Disease (IBD)
Researchers from the University of Wollongong are recruiting people with Crohn’s disease and colitis for an opportunity to test a new tool designed to support knowledge and confidence building about diet and eating with IBD.